Exercise 1: Phonon dispersion#
Estimated total CPU time: 10 min
Warning
In the following exercises, computational settings including the reciprocal space grid (tag k_grid), the basis set, and supercell size, have been chosen to allow for computation of the exercises in a limited time and within the CPU resources available during the tutorial session. Without loss of generality, these settings allow to demonstrate trends of the lattice dynamics of materials.
In the production calculations, all computational parameters should be converged.
In this exercise, you will learn:
- How to use FHI-vibes to set up and perform phonon calculations.
- How to compute phonon band structures using Phonopy.
Setting up phonon calculations#
In this tutorial, we will use CuI as an example material.
The geometry of the primitive cell of CuI needs to be relaxed before the phonon calculation (tutorials on geometry optimization). We have already done this, and we provide the relaxed geometry.in file in phonons/1_phonopy/input.
You can navigate to the directory and use the following command to check the geometry information.
Output of vibes info geometry geometry.in
[vibes] Geometry info
input geometry: CuI
Symmetry prec.: 1e-05
Number of atoms: 2
Species: Cu (1), I (1)
Periodicity: [ True True True]
Lattice:
[4.00000000e-10 3.00702569e+00 3.00702569e+00]
[3.00702569e+00 4.00000000e-10 3.00702569e+00]
[3.00702569e+00 3.00702569e+00 4.00000000e-10]
Cubicness: 0.916 (0.770)
Largest Cutoff: 1.736 AA
Bounding box rad.: 2.255 AA
Spacegroup: F-43m (216)
Wyckoff positions: 1*a, 1*c
Equivalent atoms: 1*0, 1*1
Cell lengths and angles [Å, °]:
a, b, c: 4.2526 4.2526 4.2526
α, β, γ: 60.0000 60.0000 60.0000
Volume: 54.3803 Å**3
Volume per atom: 27.1901 Å**3
Net velocity: [0. 0. 0.] Å/ps
What is the space group of this structure?
Prepare phonopy.in#
An input file phonopy.in is required for setting the calculation parameters.
Template settings for [calculator] and [phonopy] can be generated using the following command:
Settings for the FHI-aims calculator#
Here are the default parameters for calculator in your generated phonopy.in file that need to be edited:
[calculator]
name: aims
[calculator.parameters]
xc: pw-lda
[calculator.kpoints]
density: 3.5
[calculator.basissets]
default: light
[calculator.socketio]
port: 12345
The [calculator] section sets the important parameters for first principle calculations
using FHI-aims. For our example, we need to update the exchange correlation
functional from xc: pw-lda to xc: pbesol, and k-point density
from density: 3.5 to density: 2.
Full documentation on the parameters can be found in Calculator Setup in FHI-vibes documentations.
Settings for the phonon calculation#
Here are the default parameters for phonopy in your generated phonopy.in file that need to be edited:
[phonopy]
supercell_matrix: [1, 1, 1]
displacement: 0.01
is_diagonal: False
is_plusminus: auto
symprec: 1e-05
q_mesh: [45, 45, 45]
workdir: phonopy
The [phonopy] section sets the parameters for phonon calculations using Phonopy. For our example, we need to update the supercell_matrix settings, which sets the transformation matrix between the primitive cell and supercell.
As introduced in Theory: Harmonic vibrations, the force constant \(\mathbf{\Phi}_{IJ}\) considers the atom pairs in the supercell. So we need to generate a large enough supercell for describing the interatomic force constants between atom \(I\) and atom \(J\). (learn more about supercell size convergence in Exercise 3: Supercell Size Convergence from Phonons with FHI-vibes).
Following the procedure outlined in this tutorial (not done here), we find that a converged choice of the supercell matrix is \(3 \times 3 \times 3\). A command line utility is available in FHI-vibes to help determine what to enter into the supercell_matrix field for this supercell size. To use this command line utility, we need to know the total number of atoms in the supercell. For our example, CuI is in cubic phase, and the conventional cell has \(8\) atoms, so the number of atoms in the supercell is \(8 \times 3 \times 3 \times 3 = 216\). Then we can use the following command:
Output of vibes utils make-supercell -n 216 geometry.in
Find supercell for
[vibes] Geometry info
input geometry: CuI
Symmetry prec.: 1e-05
Number of atoms: 2
Species: Cu (1), I (1)
Periodicity: [ True True True]
Spacegroup: F-43m (216)
Wyckoff positions: 1*a, 1*c
Equivalent atoms: 1*0, 1*1
Settings:
Target number of atoms: 216
Supercell matrix:
python: [-3, 3, 3, 3, -3, 3, 3, 3, -3]
cmdline: -3 3 3 3 -3 3 3 3 -3
2d:
[[-3, 3, 3],
[3, -3, 3],
[3, 3, -3]]
Superlattice:
[[1.80421541e+01 1.20000019e-09 1.20000009e-09]
[1.20000019e-09 1.80421541e+01 1.19999991e-09]
[1.20000001e-09 1.20000001e-09 1.80421541e+01]]
Number of atoms: 216
Cubicness: 1.000 (1.000)
Largest Cutoff: 9.021 AA
Number of displacements: 2 (2)
Supercell written to geometry.in.supercell_216
[trajectory] .. time elapsed: 4.790s
The supercell settings can be taken from the line below Supercell matrix:, i.e. we will enter supercell_matrix: [-3, 3, 3, 3, -3, 3, 3, 3, -3].
Supercell matrix format
There are different ways one can write the supercell_matrix parameter.
Using the output of vibes utils make-supercell -n 216 geometry.in presented above, the
\(3 \times 3 \times 3\) supercell can equivalently be written as a full matrix in flattened
form [-3, 3, 3, 3, -3, 3, 3, 3, -3] or a 2-d array [[-3, 3, 3], [3, -3, 3], [3, 3, -3]].
If the matrix is diagonal, such as in the default value supercell_matrix: [1, 1, 1] this is another
option. The FHI-vibes parser accepts all these forms.
The input file phonopy.in is now complete, and you can compare your file with the example in phonons/1_phonopy/input and given below.
Example of phonopy.in
[files]
geometry: geometry.in
[calculator]
name: aims
socketio: true
[calculator.parameters]
xc: pbesol
compute_forces: true
[calculator.kpoints]
density: 2
[calculator.basissets]
Cu: light
I: light
[phonopy]
supercell_matrix: [-3, 3, 3, 3, -3, 3, 3, 3, -3]
displacement: 0.01
is_diagonal: False
is_plusminus: auto
symprec: 1e-05
q_mesh: [45, 45, 45]
workdir: phonopy
Run phonon calculation#
If you are working on a local machine, you may now perform calculations inside this folder by running
If you are working on a login node and plan to submit the work to computation clusters, you need to submit through a submission script with:
Example for the submission script on the webinar cluster
#!/bin/bash -l
# Standard output and error:
#SBATCH -o ./slurm.stdout
#SBATCH -e ./slurm.stderr
# Initial working directory:
# Standard output and error:
#SBATCH -o ./slurm.stdout
#SBATCH -e ./slurm.stderr
# Initial working directory:
#SBATCH -D ./
# Job Name:
#SBATCH -J FHI-aims
# Number of nodes and MPI tasks per node:
#SBATCH --nodes=1
# HPC7a
#SBATCH --tasks-per-node=192
#
# Wall clock limit:
#SBATCH --time=00:15:00
module purge
module load fhi-aims vibes
vibes run phonopy
In the output, you will find the following information:
...
[vibes] Compute structure 1 of 2: working
[vibes] Compute structure 1 of 2: finished.
[vibes] Compute structure 2 of 2: working
[vibes] Compute structure 2 of 2: finished.
[vibes] Close the socket
[vibes] Start postprocess.
...
Recall the finite displacements introduced in Theory: Harmonic vibrations, this output shows that \(2\) displaced supercell structures are generated and calculated to get the force constants \(\mathbf{\Phi}_{IJ}\).
The calculation should take a few minutes and you should see a
folder phonopy that contains your calculations, with resultss
collected into the file called trajectory.son. This file
contains all the necessary information from the DFT calculation, i.e.,
the structures with displaced atoms, and the according DFT forces. With
this information, FHI-vibes can "feed" Phonopy and determine the
force constants and the dynamical matrices at arbitrary \(\boldsymbol{q}\) points, from
which the phonon frequencies can be computed.
To postprocess the calculation, run the following command:
This postprocessing step calculates all the phonon properties, including force constants,
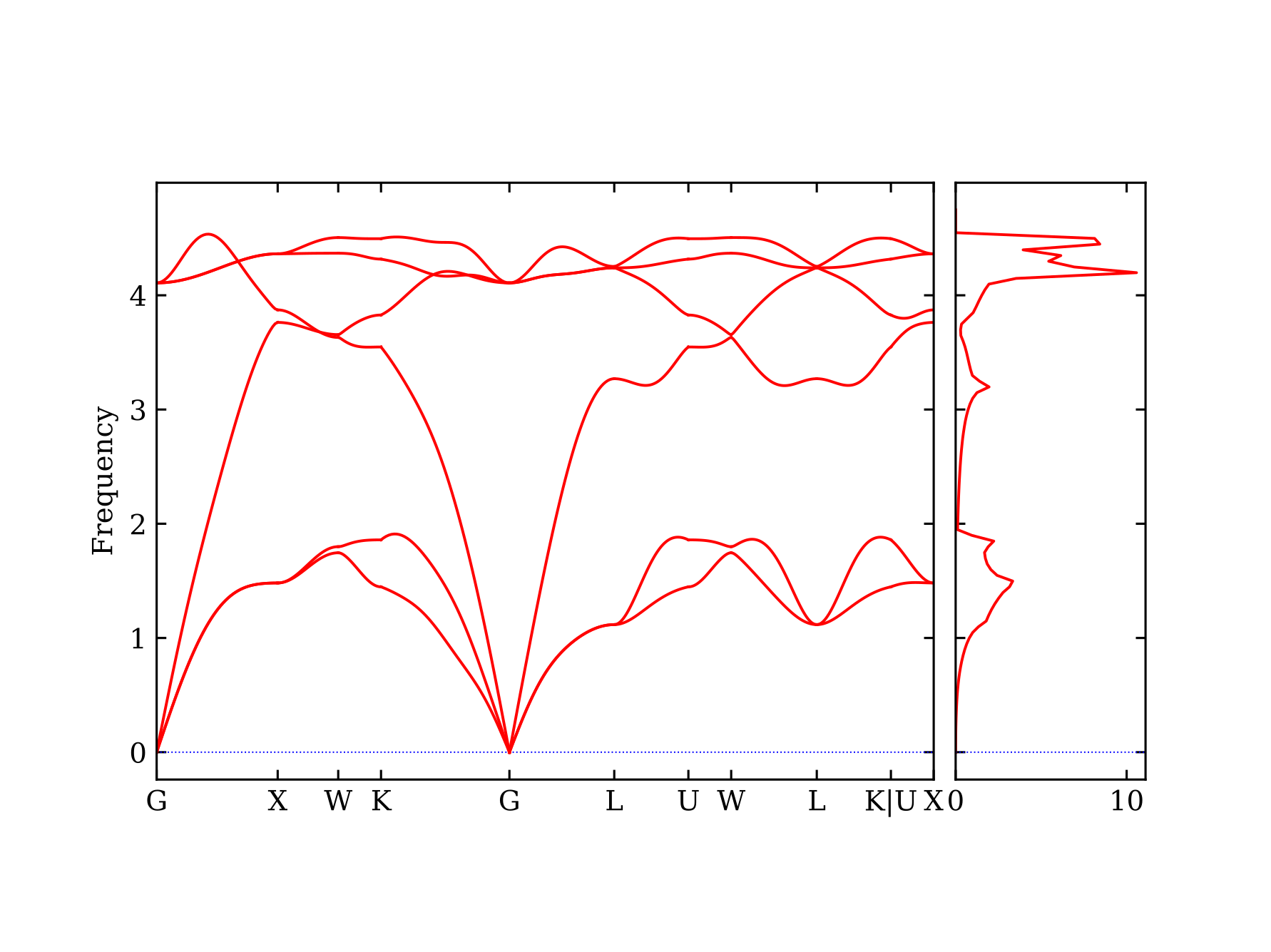
phonon dispersion, thermodynamic properties, etc. The output file will be stored in
phonopy/output. You will also find the phonon dispersion and density of states plot bandstructure_dos.pdf
in this folder, which is shown below.
Solutions#
You find all the solution to all the above exercises by clicking on the button below.